DNA Overview: how THIRD Generation Sequencing (Oxford Nanopore v 10X Genomics) is revolutionising Kennedy DNA testing! (June 2023 update)

Oxford Nanopore long read technology is squaring off against 10X Genomics linked reads
19.6.23
The raw data is in from my PacBio HiFi long read sequencing test, completing the Big Three (following earlier Oxford Nanopore and 10X Genomics tests). Initially a BAM and one VCF report were posted followed shortly after by a larger BAM which included the unmapped reads. The mapped BAM had been aligned to GRch37 (hg19) which is very old. Dante Labs have form in this regard! How inappropriate this is can be demonstrated by looking at a random DEL (deletion) SV from the VCF and converting it to GRch38 (hg38) coordinates, only to find the position has been removed from the reference genome. The first step was thus to realign the reads to hg38 which I did with minimap so as to compare with my Oxford Nanopore data. The version of minimap I used before does have a PacBio option but not one aimed at Hifi reads so after a quick run with that I upgraded to v2.26 and ran it with the map-hifi option. My chrY novel variants were then manually examined and I confirmed that all of them including the one that was only found with hg38 mapping were covered and positive, however the read depth isn't really adequate. I also manually checked a DEL on chrX I have studied before and here there was insufficient coverage to span the deletion. By contrast the nanopore and linked read tests both show it clearly.
I have also run an updated version of Sniffles to look for SVs (the Dante supplied SV VCF can be ignored as it is based on hg19). I downloaded Sniffles 2.0.7 which is now written in Python and ran it on the Pacbio data and the nanopore data for fair comparison. The results were PacBio 18913 SVs and Nanopore 44035 SVs. My other normal test using varscan I haven't bothered with yet - clearly on the sex chromosomes which I only have one copy of the findings are constrained although the NV check above indicates you can probably pick up what you need. Remember that nanopore produces 'noisy' reads with a lot of single base errors and that PacBio Hifi's big selling point is the elimination of this type of noise, an aim it has achieved in my case.
Finally just as I go to press I got an email from PacBio asking "Were you aware that low pass HiFi sequencing can deliver more comprehensive and uniform coverage than short reads can, regardless of depth?" Perhaps they are thinking of my own data? :-)
13.3.23 Keeping up with technology is vital to a successful surname DNA project and we are now trialling the latest offering using long sequencing reads. Anyone else who is interested in being a Kennedy pioneer is encouraged to get in touch. The short read market is fairly stable now with whole genome (WGS) products available from Dante Labs, Full Genomes, Nebula Genomics and YSEQ.
18.2.21 Last year the project took part in a pilot of a new 400 base pair sequencing test and this has now been officially launched by YSEQ at the cost of 335 Euros/399 USD. This test was designed to give better coverage of certain kinds of variant and we will see how good it is shortly when YFull finish their analysis of my three latest kits. The price for an entire genome sequence is very good though and reaffirms YSEQ's position as best sequencing provider not only in terms of quality but now also price.
21.10.20 Raw data was recently released from the ancient Viking DNA project (Margaryan 2019). I have downloaded all the important raw read (BAM) files and manually examined them. As usual the data quality is extremely poor but most of them can be reasonably placed on the Y tree. Samples VK95 (Iceland 10-13th CE) and VK545 (Dublin, 665-865 CE) are confirmed members of R-M222. The worst quality one is VK44 (Faroes, 16-17th C CE) which could be M222-S658 but really is lacking other markers as confirmation. The former two have been added to the M222 tree on YFull.com and added to the M222 project there. The Dublin sample being by far the oldest is the most interesting. However as usual we need a lot more samples and they need to cover Scotland as well as Ireland to really make sense of the past and modern distribution. Ayrshire and Perthshire especially, please!
30.9.20 I have now evaluated nine different sequencing tests - a mix of targetted Y, short, long and linked read whole genome tests. In addition I have also carried out six different SNP array/'chip' tests as well as used microsatellite (legacy STR) tests from six different vendors. The total number of labs utilised is sixteeen! It is doubtful anyone else has carried out such a breadth of testing across the market and maintains a commitment to continue doing so as new options arise. Out of these, the YSEQ.net WGS (whole genome) test remains the flagship product and the mainstay of the Kennedy DNA project. It is head and shoulders above all the others in terms of quality, turnaround and the scope of the analysis reports. In addition all data inluding a BAM file of the entire genome are immediately available for download with the reports - some companies charge you extra for the raw data and don't even reveal the fee at the time you order the actual test! The new Italian startup Dante Labs continue their activities but sadly their customer service remains erratic, although they have offered both short and long read nanopore tests previously. As of autumn 2020 they are only offering short read tests but I remain hopeful they will re-enter the nanopore market in due course.
2.3.20 Updated the varscan spreadsheet with my Sano Genetics BAM file analysis, this scored 16/17 on the FGC NVs just missing FGC5856 due to insufficient depth. The default threshold in varscan is 8 reads and this position only got 5.
12.2.20 Last year was a very successful one for the project with 30 male Kennedys being DNA sequenced and uploaded to the Kennedy project tree at YFull. Another test has just been kicked off at the lab. The evaluation of the marketplace continues with pilot test data now in from both Chronomics and Sano Genetics.
15.11.2019
The BAM file for my Dante short read WGS has now been delivered online and was downloaded yesterday and forwarded to YFull.com. Two FGC novel variants that were not previously called in the Dante VCF - FGC5870 and FGC5873 - were confirmed as derived in the raw data. This should serve as a warning to anyone being told by *any* vendor that you don't need a BAM file! Without the actual data you are at the mercy of the arbitary variant calling protocols of the lab. There were also links to download the paired end FASTQ files but the files were not accessible so I extracted them from the BAM using Picard for local realignment. The mapping to hg38 has been done using minimap2 as with the Dante long read data and SNPs called as before with varscan. The variant calling scored 100% although FGC5865 was called as 0/1 due to the inclusion of 8 MAPQ=0 reads. I have zero MAPQ reads toggled off in IGV so didn't see that on the initial manual inspection. I've updated my varscan spreadsheet.6.9.2019 update.
I attended the three day Genome Science 2019 conference in Edinburgh. Of particular interest were talks by Clive Brown, the chief technology officer at Oxford Nanopore, whose firm I tested with earlier this year (too brief unfortunately); Matt Loose who holds the 'world record' for the longest ever sequencing read at 2.3Mb; and Sergey Koren on the 'Telomere to Telomere' (T2T) project which recently closed all the gaps on the human X chromosome including the centromere and telomeres. Wrapping up the final day was a fascinating talk by Dr. Kirsten Bos of the Max Planck Institute on the genetic history of the bubonic plague. This page covers phases 2 and 3 of my Kennedy DNA study, historical notes on phase 1 and some haplogroup notes about my own Y chromosome group (M222, FGC4077 and FGC5856), and finally some remarks on the ever popular autosomal tests. Kennedy DNA study Phase 3 (2019+) Phase 3 of the project is examining third generation long read sequencing technologies from Oxford Nanopore and PacBio, along with the 10X Genomics Chromium Linked Read system which adds barcoding to traditional second generation sequencing. As of May 2019 these tests are all available for exploration but are still on the expensive side and their benefits in genetic ancestry and surname studies are still to be established. For the 10X Genomics Chromium system, sold by Full Genomes, I have now received analysis, BAM and FASTQ; the analysis consisted of output from the 10X Genomics LongRanger pipeline as well as FGC's own analysis. For the Oxford Nanopore test, conducted by the newly certified Dante Labs facility in l'Aquila Italy, I have so far only been given a FASTQ file. This means it has raw sequencing reads but they are not aligned to a reference genome. I conducted my own alignment using minimap2, an aligner designed to handle these kinds of long reads. By May 25th I had run my own LongRanger WGS pipeline on the 10X FASTQ files. This gave me amongst other things the loupe file the lab had declined to forward. Loupe is a visualization tool for the Long Ranger pipeline output. The first thing I found opening up my loupe file was a big red banner at the top announcing 'The analysis detected some serious issues with your sequencing run'. Clicking on the link informed me of (a) Low linked-reads per molecule value 7.0 Ideal > 13. Secondly Low Input DNA Molecule length 13794.1, ideal > 40000... The pipeline gives the user the choice of GATK or FreeBayes for variant calling so I selected the latter for comparison with the lab supplied GATK analysis...Variant calling using varscan has now been performed on the YSEQ, ONT and 10X test BAM files and the results summarised in a spreadsheet.The most striking feature of the comparison is the high false positive rate for INDELS in the Oxford Nanopore test data. This sort of issue is typical of third generation sequencing, see for example the discussion by Magi (2016). There is also now (4.6.19) a summary of the total number of Y chromosome SNPs reported and a tally of how many of my FGC novel variants each test found.
In order to further compare the three tests the BAMs have all been put through Alfred for QC. One helpful feature of Alfred is the online viewer for the analysis metadata and the output for the three BAMs under discussion (ONT, 10X and YSEQ) can be uploaded for comparison by interested readers (submit each .json.gz file in turn then click 'Launch Analysis'). Note the different lengths for the 10X barcoded read1/2, but Alfred is not barcode aware and is only reporting the underlying short reads not the linked reads anyway (NB it does report the phasing blocks though). The Loupe report on my news page shows a histogram of the linked read lengths. Incidentally YSEQ are now trialling 250bp short reads as well as exploring nanopore sequencing themselves.
Sniffles structural variant (SV) calling has now (5.6.19) been conducted on the nanopore reads - despite the author stating that only BAMs aligned with bwa-mem or NGMLR are accepted (output referred to as sniffONT.vcf). The run-time was around an hour after preparing the BAM file with MD tags. This variant caller reports on Deletions (14419), Inserts (12468), Inversions (106), Duplications (94), Inverted Duplications (8) and Translocations (0). These need downstream analysis to determine if they are previously recorded in dbVar - you can download tsv files of all the variant types - and to determine if they contains false positives. Analysis can include comparison in BEDtools/mySQL or visualising in IGV using the BED files at the above link. Long Ranger has already reported on deletions and larger scale SVs from my linked reads data.
The Long Ranger pipeline produces a number of reports that may not be familiar. The first to examine is phased_variants.vcf, a 3.5Gb report. Genetic genealogists who have done a Y chromosome targetted sequencing test may have seen a VCF (variant call) report before. For a whole genome of course you will record two alleles at each base for the autosomes but the more significant difference is that most of the variants (97% of my sample according to Long Ranger) have been 'phased' ie divided up into parental haplotypes. This can be achieved wherever the sample has heterozygous alleles, locations with one base from one parent and another from the other eg AG. This enables the construction of phasing blocks split up by homozygous alleles. Alleles that are phased are denoted by the | separator (eg 0|1 rather than 0/1). Linked read phase blocks can reach impressive lengths, my longest one is about 2Mb and the N50 length is 262kb. There is further relevant data in the BAM file, particularly the Molecule Identifier (MI) tag as well as the original raw barcodes as the VCF barcodes have been error corrected.
If you have the required data you can cross-check this phased data with results for known relatives who have done autosomal chip tests, however you need them to have shared all their raw data with you. Another 10X customer has been able to do this but it probably will not be possible for me. Phasing in itself isn't new in genetic genealogy of course although typically it has been done using information derived from parents. There are some useful discussions on it by both Ann Turner and Blaine Bettinger in the recent book 'Advanced Genetic Genealogy: Techniques and Case Studies' edited by Debbie Parker Wayne (not covering long and linked reads though).10.6.19 Of the three remaining reports two relate to large scale structural variants (SVs) and one reports on deletions - dels.vcf. This last report emerged from the Long Ranger analyze_sv_calls pipeline stage and my lab supplied version contains 4728 rows covering the autosomes as well as the X and Y chromosome. The del lengths range from 41bp to ~26kbp. The longest of these 'deletions' seems to be partially supported by a Long Ranger phase block at its terminus (chr11:13821023) but confirmation in the nanopore data is lacking. Unfortunately IGV doesn't have the fullness of support for linked reads that Loupe does whilst the latter won't display non-10X data. There is a screenshot of a chrX deletion on the news page with cross-lab support and chr3:61141516 is a ~1k deletion which is also clearly visible in the nanopore data but in contrast to the X example, not so clear in the YSEQ short read data due to it being a heterozygous deletion. Even a sharp drop off in read depth for a deletion on one allele can be overlooked if the coverage in the area was fluctuating anyway.
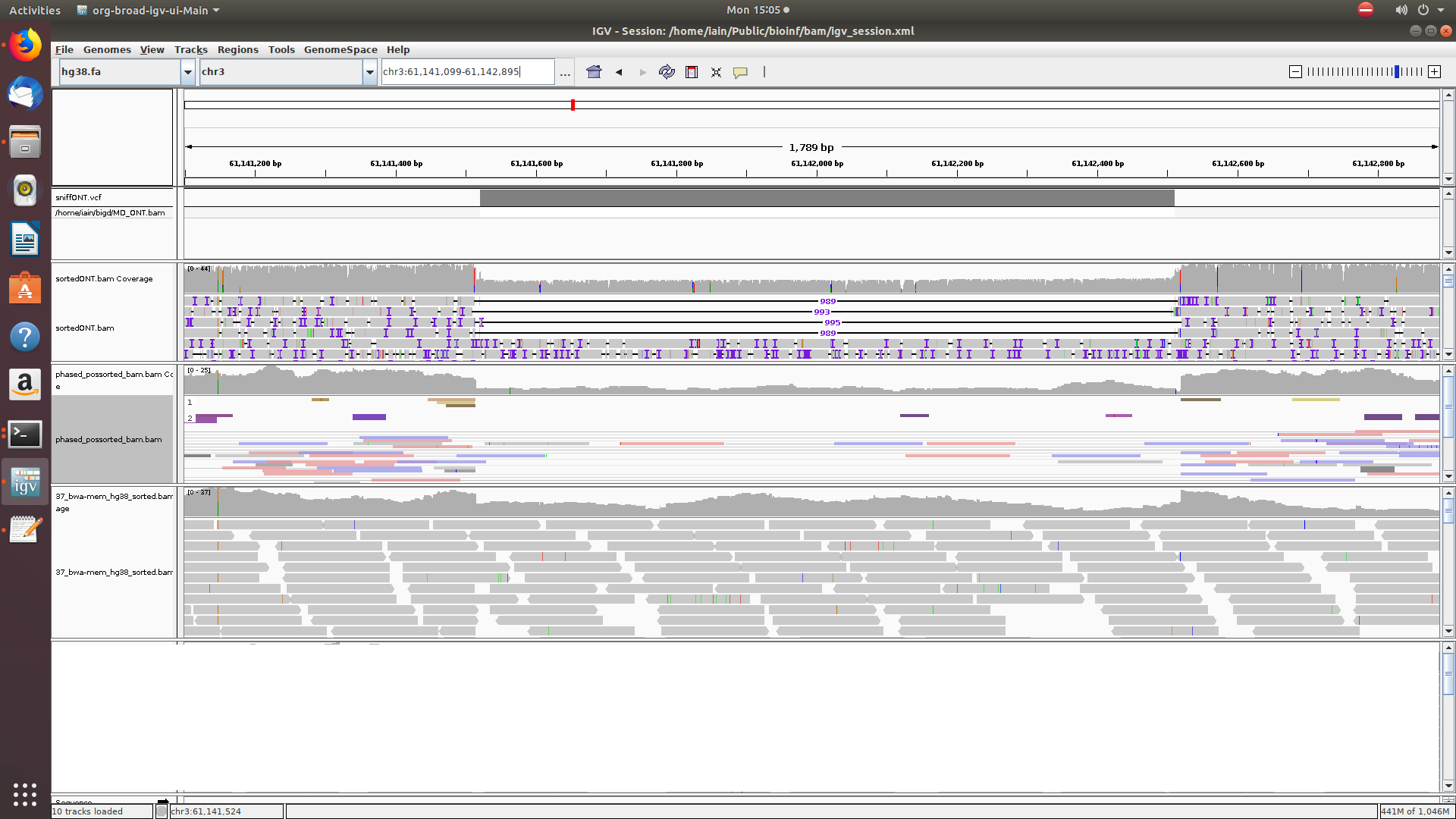
A heterozygous deletion viewed in IGV with 10X (middle pane) assigning the deletion to haplotype 1 after phasing.
The length distribution of the entries in dels.vcf is highly skewed with one third falling in the ~300bp range. I believe these are mostly Alu elements, a type of short interspersed nuclear element (SINE). There is a smaller peak visible around the 6kbp mark where the longer LINE elements appear. Both these identifications are now confirmed by cross-checking with the UCSC repeats track. The double-peaking length distribution is typical of long read sequencing as illustrated in the recent SV benchmark paper from the Genome in a Bottle project. Alus have been used in human phylogeny studies in the past, and Y chromosome haplogroup DE is defined with the 'YAP' Alu.
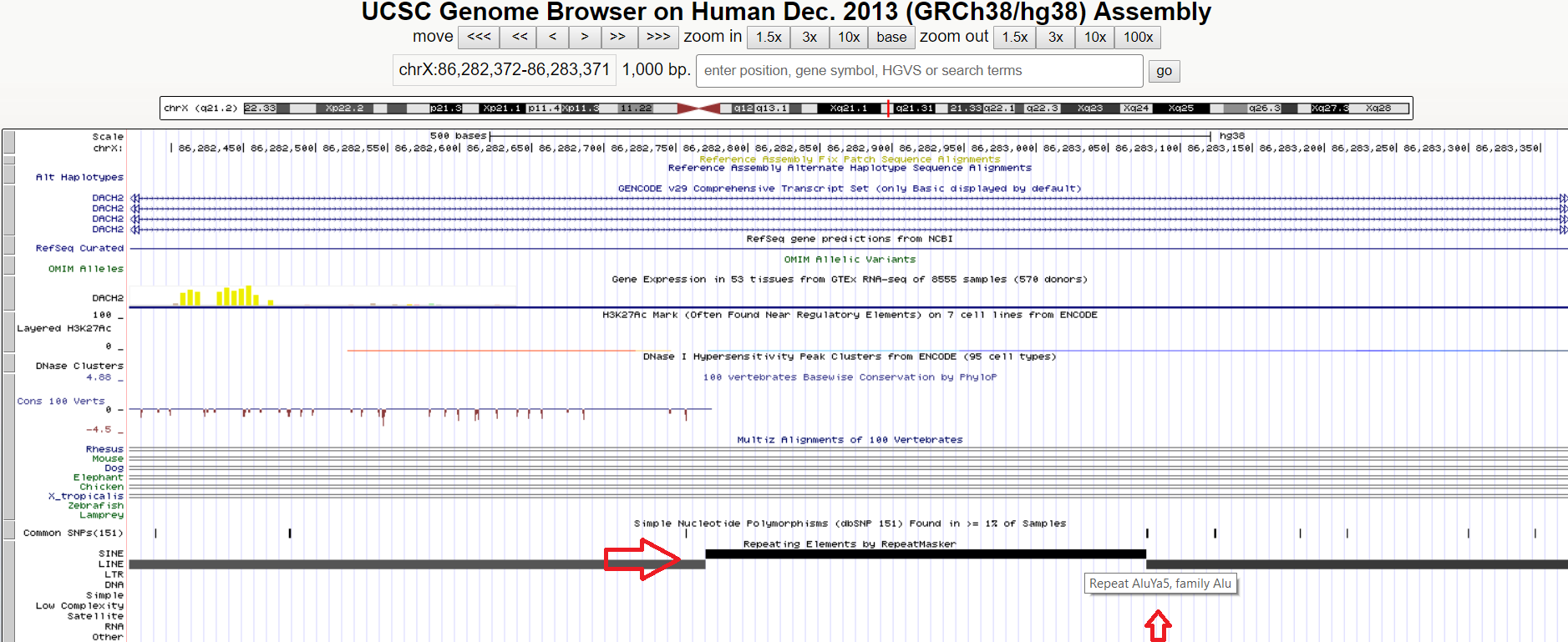
An Alu element viewed at UCSC, showing as a 312bp deletion in my Sniffles nanopore report.
17.6.19 The remaining report relates to large structural variants - large_svs.vcf. These get broken down into candidate and real calls in Loupe and in a pair of .bedpe files. The size of variant reported ranges from ~23kbp up to 33Mb. Analysing these reports requires a mixture of visualization in Loupe/IGV, cross checking against my Sniffles report from the Nanopore testing (manually and/or using bcftools-isec and bedtools-intersect) and finally comparing with other samples to see if they are novel or not. So far I have only been able to confirm deletions in the 30-40kbp length range (record is 49kb so far with a possible 155kb apparent but not called by Sniffles). It is worth noting that the examples 10X show are also modest in size, a homozygous 50kb deletion and a 160kb heterzygous one. Many of the longer variants reported appear to be spurious and include such unlikely events as the deletion of the PAR region and even some centromeres.
The following table provides a summary of the deletion entries in the three VCF files. There are a small number of other variant types which will be looked at in due course.
| Technology | File | Rows | Min | Max |
|---|---|---|---|---|
| 10X | dels.vcf | 4977 | 41bp | 28kbp |
| 10X | large_svs.vcf | 346 | 30kbp | 33.1Mbp |
| Oxford Nanopore | sniffONT.vcf | 14419 | 31bp | 11.8Mbp |
23.6.19 It is also possible to compare the personal VCF reports with the composite SV callset from the 1000 Genomes Project which brings the advantage of not only categorising all the Alu and LINE elements but also calculated allele frequencies (AF) on both a world and regional basis, along with genotypes for each project member. The callset contains some 13k Alus (of which about 10% are 'deletes' or SVTYPE=DEL_ALU) and around 3k LINE elements. The intersecting calls from my personal VCFs are 1184 from 10X dels.vcf and 1485 from sniffONT.vcf of which around a third of each are Alus. There are no overlapping calls from the 10X large_svs.vcf report.
27.6.19 Y chromosome phylogeny. Both runs of the Long Ranger pipeline have produced an identical list of 20 SVs on the Y chromosome (length 44-9266bp). By contrast Sniffles has reported 67 deletion SVs on the Y chromosome ranging in length from 31-2051bp (some like to maintain 50bp minimum to be called a structural variant). There does not appear to be any overlap between the two variant lists. Many are not located precisely by the calling algorithm, for example Sniffles only classified 30 out of 67 as PRECISE location. As mentioned above it is possible to do fuzzy matching using bedtools if you are prepared to pick an arbitrary percentage overlap figure (eg 95%). Many of the Sniffles variants called are in poor regions of the chromosome (eg CEN, DYZ19). To date nothing has been found in these two reports that is both a true positive and of phylogenetic significance although work is still ongoing. There are variant reports produced in-house by Full Genomes (in addition to the supplied Long Ranger output) which also needs more analysis.
3.7.19 The low-pass WGS from Full Genomes back in 2015 included a lab report for Copy Number Variants (CNVs) from the Control-FreeC tool. Given the low read depth (average 2x) in this test it may be unfair to expect much. For example amongst the 70 gains and 32 losses it reports a CNV gain in the DUSP22 gene on chr6 but re-running Control-FreeC on the new nanopore data doesn't produce a matching report entry - although it is possible to make out a modest jump up in read depth at the supposed start breakpoint.
Some early comments on the 10X can be found in my sequencing summary report but the bulk of the output from both tests has still to be analysed. These tests are more complex to deal with than traditional WGS tests and note in particular that nanopore testing on its own is unlikely to be a first choice for Y SNP discovery. They are very powerful in other ways including the potential phasing of my genome into paternal and maternal haplotypes which can either be analysed from the BAM and VCF files or visualized in the loupe browser.
The project will also explore the use of Deep Learning GPU based software for genomic data analysis. Kennedy DNA study Phase 2 (2013-2019+) The seeds for phase 2 were probably sown in 2012 with the publication of the citizen science paper by Rocca et al. on mass SNP discovery and Dr. J Wilson's sequencing of two R-M222 males, resulting in the breakthrough 2013 SNP array test called 'Chromo2'. This affordable test for the first time enabled surname projects to switch over to a SNP-first approach. Chromo2 was rapidly overtaken by more flexible SNP test panels developed by Dr. T. Krahn, one of Rocca's co-authors, made available via his 'YSEQ' lab in Berlin. The advantage of the SNP/next-generation sequencing method is simply that the rates of false positives are virtually zero - in fact since 2013 there has not been a single false positive in the Kennedy project. Professor Mark Jobling* (2017) writes "SNPs define stable haplotypes, known as haplogroups, which can be used to build a robust phylogeny using the principle of maximum parsimony". Depending on budget and whether funding is coming from the project, testing usually involves the YSEQ lab for single and fixed panel SNP testing and either YSEQ or Full Genomes Inc. for the more advanced sequencing (sometimes loosely referred to as 'SNP discovery' testing).Further background on the explosive growth in Y-SNPs can be found in the article 'The Y-Chromosome Tree Bursts into Life'; this was published in December 2014 since which time the tree has grown in size by a further entire order of magnitude.
Most of the branches studied during Phase 1 have now been retested using SNP testing methods, as well as using the latter for new samples. With SNP results it is much easier to draw unambiguous 'trees' (cladograms) showing the exact relationships of all the Kennedy branches. The R-M222 part of the overall Kennedy results tree is public on this site and shares results with general M222 men due to surname-haplogroup collaboration efforts (see the M222 secton below). Men sequenced by the project who lie outside M222 can be seen on the YFull Y chromosome tree. All the Scottish and Ulster branch discoveries were made by my project. Dr. Thomas Krahn identified the core marker for the Tipperary ('Dalcassian') O'Kennedys in 2009.The project will cover the costs of testing if a male Kennedy is available with a proven paper trail to a rural parish in Scotland or Ireland. Men not qualifying under those conditions are of course more than welcome to test under direction of the project. As of October 2018 funding may be extended to cover the more detailed sequencing test on a 'while stocks last' basis. This free test is worth over 600 Euros. There are currently 19 kits sequenced or being sequenced as of mid March 2019! A choice of European or American lab is available. The latest kits have benefited by a YSEQ pilot using 250 base pair reads.
IMPORTANT: ALL KENNEDY BRANCHES (SNPs, HAPLOGROUPS, GEOGRAPHIC ORIGINS) ARE OF EQUAL INTEREST TO THE PROJECT!! From the Dingle peninsula to the Shetland Isles. Kennedy DNA study Phase 1 (2004-2012) The depracated Y-STR testing method was commercialized back in 2000, using techniques explored by academics Dr. Bryan Sykes and Dr. Mark Thomas. Their papers on the Sykes surname and the Cohanim priesthood set the scene for what ultimately turned out to be a flawed matching system which in some instances could have false positive rates exceeding 90%. My own Y-STR testing commenced in 2004 and from 2006 to early 2013 I tested considerable numbers of male Kennedys using this method. This data now has to be considered obsolete. The M222, FGC4077 and FGC5856 haplogroups (2006-2019+) I was a first day tester of the M222 SNP in early 2006 when Drs. James Wilson and David Faux first put it on sale at the sadly missed Ethnoancestry lab. Because of this I divide my time between the Kennedy surname project and M222 research - of course my interest grows as I near my own sub-branch M222-FGC4077 and then FGC5856. Kennedys occur in no less than EIGHT sub-branches of M222. This is why I publish a joint chart of M222 and Kennedy data. This tree extends upwards and sideways but the non-M222 sections are not online at the time of writing. The chart is on pdf format and can be text searched for surnames of interest eg Kennedy! So what exactly is this chart? Originally it was designed as a CHROMO2 RESULTS TRACKER. Therefore the tree it started off with was of course the Dr. Wilson M222 tree published during the rollout of Chromo2. Within weeks my own sequencing data had determined that FGC4077 was the branch accounting for most of the unassigned testers on Chromo2 and so this branch was added to the tree. As I identified new branches these and the test surnames were also added. At all times branches were only added after I had proven them to my satisfaction based on raw data. Apart from the original Wilson markers (which numbered around 24) it was my own work and not copied from anyone else's tree. This research continued until mid-2015 when I reduced my direct work and started to rely more on the analysis work of YFull.com who do professional BAM data analysis. I co-admin the YFull M222 project with Aidan Byrne. A review of the history of M222 research from 1999 onwards can be read here (the August 2019 update covers breaking news on the first M222 ancient DNA). Following my own sequencing and the discovery that the FGC4077 branch represented in excess of 10% of all M222 men, informal group discussions led to a suggestion to create a mailing list which was set up by Gerry Hoy, I act as the admin/moderator. Results are shared via my M222 chart and the YFull R-Y3454 tree their name for the group). This group covers everything at or below FGC4077, FGC4078, FGC4087, A725, FGC12948, FGC5856 and A361 sub-branches and so on below. If you have a positive result for one of these, please contact me to join the group. The M222 SNP chart including the M222 Kennedys is here. I almost forgot - what about Autosomal testing?! In the last few years affordable tests from Ancestry.com and 23andMe have become household names and have taken over the bulk of the market share. Both have multi-million sized databases with Ancestry recently announcing they had tested 10+ million men and women (now shipped out 14 million kits!). Most of the markers they test are on the autosomes chromosomes 1 to 22 and the X chromosome. Confusingly though, they do have a small selection of Y chromosome markers as well as mitochondrial (mtDNA) markers. Although they have improved they are not comparable with the detailed and very current Y SNP test panels from YSEQ. The tests are hence good for finding close cousins on all tree branches but poor for deep examination of male kindreds. I continue to urge takers of these two tests to get their raw data uploaded to the comparison site GEDMatch (or Genesis-GEDMatch for the latest 23andMe v5 test). The old GEDMatch data has now been migrated to the Genesis version of the site. If you are a Kennedy, drop me a line with your GEDMatch kit number. We can then discuss whether the raw data contains any Y SNP markers of use. I have done both the Ancestry and 23andMe tests as well as the new LivingDNA test from England. The latter is similar to the first two but the matching database has yet to launch.* Professor Mark Jobling is a leading UK geneticist and was the Y-SNP consultant to LivingDNA on their new SNP array test.
For further information please contact the project.